The pharmaceutical industry has been through a very active merger and acquisition period and as a consequence,process changes abound as manufacturers take actions to make the most efficient use of new assets. The industry movement from stainless steel to single-use pre-sterilised systems and components is another drivingforce for process changes.Other sources of change include the transfer of processes, scale-up, moving of facilities, product line extensions and formulation changes and the preparation for and response to regulatory inspections.To remain cGMP compliant in a rapidly evolving environment, it is imperative that a robust change control program is in place to evaluate the risk and impact of process changes on a validated process or system. Sterile filtration of a drug product is a highly critical process that protects patient safety, the following key points should prove helpful when evaluating the impact of process change on the validated state.
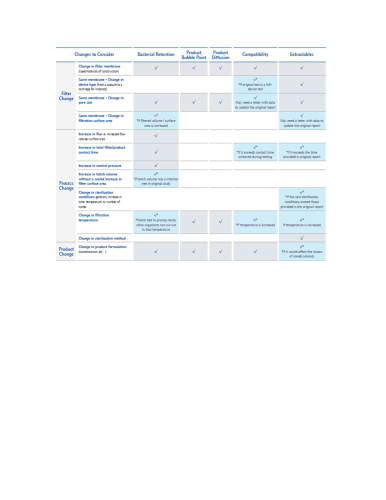
Changes that require revalidation
Filter validation processes assess filter performance with a given product and in a given process.“When designing the validation protocol, it is important to address the effect of the extremes of processing factors on the filter capability to produce sterile effluent. Filter validation should be conducted using the worst-case conditions, such as maximum filter use time and pressure.” [1]Any drug product, filter or process change should trigger the need for a revalidation assessment of a sterile filtration process (see Table 1).
Product change
Any changes to the product formulation, including changes in active pharmaceutical ingredient (API) or excipient concentration and pH, will require revalidation if the product variables have not been bracketed by previous testing. Bacterial retention testing, integrity testing and compatibility testing will have to be performed again. As for extractables, a new study should be conducted only if the change would affect the choice of model solvents or if aleachables study was conducted.
Filter change
If the membrane and membrane area are the same and only the device changes (from a cartridge to a capsule for example), only the extractables study and the compatibility test should be repeated. As for changes in the filter membrane type, these will require all filter validation studies to be repeated.The same applies when the filter membrane type is the same but the pore size changes, with the exception of compatibility which does not necessarily require a new study. When there is a change in filter size and thus in filtration surface area, bacterial retention testing might have to be repeated if there is a subsequent increase in filtered volume per surface area, pressure, flow rate, or filtration time.
Process change
Per the 2008 Revised PDA (Parenteral Drug Association) Technical Report 26, when the validated temperature range is exceeded, the entire filter validation should be repeated. Because sterilisation conditions are impacting the level of extractables, changes in the sterilisation method or increases in sterilisation conditions such as time and temperature will require revalidation [2].For example:• Keeping all other variables such as filter and product constant, increases in key process parameters such as flux (flow rate/surface area), pressure, filter/product contact time, or filtered volume/surface area which exceed the conditions of the previous testing will require bacterial retention revalidation. Compatibility and extractables studies might also need to be repeated in specific cases.• When the validated temperature range is exceeded, the entire filter validation should be repeated. Because sterilisation conditions are impacting the level of extractables, changes in the sterilisation method or increases in sterilisation conditions such as time and temperature will require revalidation.
Methodology
It is important to periodically review filter validations especially when there are proposed process changes or excursions. There should be a change control programme which triggers a review of validation documentation. Riskassessment should be done to identify the systems and processes that are critical to the manufacture of safe and efficacious drugs and have the highest chance of a change to the validation status.A best practice would be to perform a regular periodic review of the validated state of the sterile filtration process. Autoclaves and steam-in-place processes, for instance, are typically on an annual revalidation cycle while sterilisation by filtration is not. However, changes to the filtration processcan change the state of the validation and go unnoticed. A few common examples where filtration process changes can have unnoticed implications on the validated state include the following:• An increase in filling line speed that then exceeds the maximum flux (flow rate/surface area) achieved during filter validation of microbial retention. Flux can be a worstcase parameter that is validated during a bacterial retention study. A manufacturer that boosts efficiency by increasing filling line speed or by expanding a product to a largerdose volume could without notice exceed the qualified parameters of its sterile filter validation.• A move from an autoclavable filter capsule to a pre-sterilised gamma irradiated filter capsule would impact the extractables profile. This type of change is common as drug manufacturers utilise more presterilised components and devices. This type of change can also go unnoticed since the base polymers of the material of construction may remain the same but for a gamma sterilised device would likely contain additives for proper gamma stability of the materials.• Post-use integrity testing of a sterilising grade filter used for the filtration of a drug product is a GMP requirement. If the postuse integrity test is conducted with the drug product as the wetting fluid it can also be a good way to maximise product recovery and yield by simultaneously blowing down the filter. Drug manufacturers often implement steps like this through continuous process improvement efforts. However, implementation of this practice subjects the in process filter up to the filter’s bubble point pressure and could very likely exceed the maximum differential pressure that was achieved during bacterial retention validation.
Key questions?
The following are questions to ask and suggestions on how to evaluate your current filter validation status.• Identify all currently manufactured products.• Have they been validated? If so, review older validations against the current process conditions.• Have there been filtration process changes?• Has the product formulation changed since the validation, as changes in formulation can be seen as ‘new’ products?• Identify the products going into Phase 2 or 3 clinical trials and those being submitted for NDA or ANDA.• Have all filtration processes been validated?Finally a strong validation service partner should be able to offer valuable expertise and service in assessing the impact of change on the validated state.
References
[1] Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing, Current Good Manufacturing Practice. Rockville, MD: U.S. Dept. of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (2004), p. 28, paragraph 2.[2] PDA Journal of Pharmceutical Science and Technology, Technical Report No. 26, Sterilizing Filtration of Liquids. Volume 62, No. S-5 (2008), pp.9, section 4.1.1.
Contact:Ross W. AcucenaRegulatory Consultant, Services and SolutionsProcess Solutions Business UnitEMD Millipore



